
Molybdenum cofactor deficiency type A
At a Glance
 It is a chronic, life-threatening condition that was previously untreatable but can now be managed with a specific medication approved for type A.
It is a chronic, life-threatening condition that was previously untreatable but can now be managed with a specific medication approved for type A.










How It Affects You
Molybdenum cofactor deficiency type A is a rare genetic metabolic disorder that prevents the body from breaking down certain waste products, specifically sulfites. This inability leads to a rapid accumulation of toxic chemicals that causes progressive and severe damage to the central nervous system.
- Toxic sulfite buildup destroys brain tissue, leading to swelling and deterioration.
- Intractable seizures and muscle rigidity or floppiness affect the entire body.
- The condition can cause widespread developmental delays and life-threatening complications if not treated immediately.
Causes and Risk Factors
Genetic Causes and Biological Mechanisms
Molybdenum cofactor deficiency type A is caused by mutations in the MOCS1 gene. This gene provides instructions for making a compound called cyclic pyranopterin monophosphate (cPMP), which is the first step in creating the molybdenum cofactor. This cofactor is essential for the function of three specific enzymes in the body: sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase. The most critical failure is the lack of sulfite oxidase, which is responsible for breaking down sulfites (a type of chemical found in the body and some foods). When this enzyme fails, toxic levels of sulfites accumulate in the body, while uric acid production decreases. This chemical imbalance acts as a neurotoxin, rapidly destroying brain cells and causing swelling (edema) and tissue loss (atrophy) in the central nervous system.
Inheritance and Risk Factors
The primary risk factor is family history, as the condition follows an autosomal recessive inheritance pattern. This means a child must inherit two copies of the non-working gene—one from each parent—to develop the disease. Parents who each carry one copy of the mutated gene have a 25% chance of having an affected child with each pregnancy. Carriers typically do not show any symptoms.
Prevention and Screening
There are currently no methods to prevent the initial genetic mutation that causes the disorder. However, for families with a known history of the condition, genetic counseling and prenatal testing are available to understand the risks. Newborn screening panels in many regions do not routinely check for this specific condition, so prevention of damage relies on rapid identification of symptoms after birth. Preventing the progression of brain damage depends entirely on how quickly medical treatment is started after the first signs appear.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
Symptoms usually begin within the first few days of life, although in rare cases they may appear later. The most characteristic early sign is intractable seizures (seizures that do not stop with standard medication). Other common physical signs include:
- Feeding difficulties: Infants may have trouble latching, sucking, or swallowing.
- Exaggerated startle response: Babies may react intensely to noise or touch.
- Abnormal muscle tone: This can present as floppiness (hypotonia) or stiffness and spasticity (hypertonia), often affecting the arms and legs.
- Facial features: Over time, some children develop distinctive facial features, such as a long face, puffy cheeks, and widely spaced eyes.
- Eye abnormalities: Dislocation of the lens in the eye (ectopia lentis) can occur as the disease progresses.
- Microcephaly: The head size may be smaller than average due to poor brain growth.
Diagnostic Tests and Exams
Clinicians suspect this condition in newborns who have seizures that are difficult to control. Diagnosis is confirmed through a combination of biochemical tests and genetics:
- Urine and Blood Tests: A simple urine dipstick test can show high levels of sulfites. Detailed lab work will reveal low levels of uric acid in the blood and high levels of xanthine and S-sulfocysteine in the urine.
- Genetic Testing: Molecular genetic testing identifies mutations in the MOCS1 gene, which confirms the diagnosis of type A specifically.
- MRI Imaging: Brain scans often show a specific pattern of damage, including swelling and fluid-filled cysts, which helps distinguish it from other causes of newborn seizures like hypoxic-ischemic encephalopathy (HIE).
Differential Diagnosis
Doctors must distinguish this condition from Hypoxic-Ischemic Encephalopathy (lack of oxygen at birth), other metabolic disorders, and hereditary hyperekplexia (startle disease), as the symptoms can look very similar initially.
Treatment and Management
Medication and Targeted Therapy
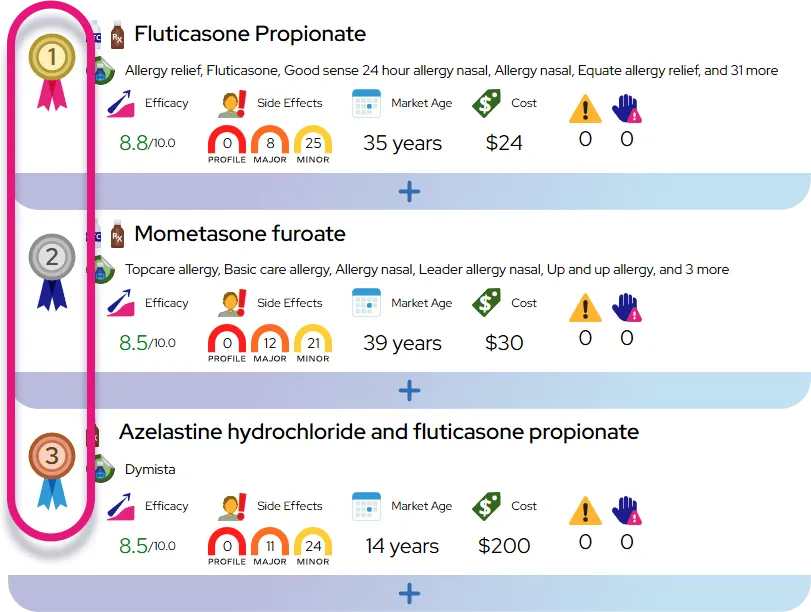
The primary treatment for Molybdenum cofactor deficiency type A is fosdenopterin (brand name Nulibry). Approved by the FDA in 2021, this is a substrate replacement therapy that provides the body with the missing cPMP compound. By injecting this medication intravenously (IV) daily, the body can produce the necessary molybdenum cofactor, allowing the sulfite oxidase enzyme to work and reducing toxic sulfite levels. This treatment is life-saving and is most effective when started as soon as possible, before permanent brain damage occurs.
Symptom Management
Supportive care is also required to manage the complications of the disease:
- Seizure Management: While fosdenopterin reduces seizure risk, anti-seizure medications may still be needed initially.
- Nutritional Support: Feeding tubes (gastrostomy tubes) may be necessary if the child has difficulty swallowing to ensure proper nutrition.
- Physical and Occupational Therapy: These therapies help manage muscle stiffness (spasticity) and support motor development.
- Respiratory Support: Some infants may require breathing assistance or oxygen if muscle control is poor.
When to See a Doctor
For parents of a newborn, immediate emergency care is required if the baby experiences seizures, repetitive rhythmic movements, or extreme lethargy. If a child has already been diagnosed, parents should seek medical attention if:
- Seizures return or change in pattern.
- There are signs of infection, such as fever or vomiting, which can stress the body's metabolism.
- The child has difficulty breathing or feeding.
Severity and Prognosis
Severity and Disease Course
Molybdenum cofactor deficiency type A is considered a severe and critical medical condition. Without targeted treatment, the disease follows a rapid, progressive course. Toxic substances accumulate daily, causing irreversible destruction of brain tissue (encephalopathy). This leads to profound intellectual disability, inability to move voluntarily (spastic quadriplegia), and severe health complications.
Life Expectancy and Mortality
Historically, the prognosis was grim. Infants who did not receive the newer targeted therapy typically had a median survival of about 3 to 4 years, with death often caused by respiratory infections or complications from intractable seizures. However, the introduction of fosdenopterin has fundamentally changed this outlook. Clinical studies have shown that treated patients have a significantly higher survival rate compared to untreated historical cases.
Prognosis Factors
The most critical factor influencing the long-term outlook is the timing of treatment. Because brain damage caused by sulfite toxicity is permanent, treatment started immediately after birth (or even before symptoms appear in siblings of known cases) can lead to near-normal or significantly improved development. If treatment is delayed until after significant brain injury has occurred, the medication improves survival and alertness but cannot reverse existing disability.
Impact on Daily Life
Daily Activities and Caregiving
Caring for a child with this condition often requires 24-hour supervision and support. Daily life may revolve around a strict medication schedule (daily infusions), feeding sessions (possibly via tube), and managing medical appointments. Children may have significant physical limitations requiring wheelchairs or adaptive equipment. Sleep patterns may be disrupted due to neurological issues.
Support and Coping
Families often face high emotional and financial stress. Connecting with rare disease advocacy groups and connecting with other families affected by metabolic disorders can provide essential emotional support. Respite care is often recommended to prevent caregiver burnout.
Questions to Ask Your Healthcare Provider
- Is my child eligible for the targeted therapy fosdenopterin, and how do we access it?
- How do we administer the daily intravenous injections at home?
- What specific signs of metabolic crisis or infection should we watch for?
- What is the best way to manage feeding and nutrition to prevent complications?
- Are there clinical trials or registries for this condition that we should join?
- What genetic counseling resources are available for our future family planning?
Common Questions and Answers
Q: Is Molybdenum cofactor deficiency type A curable?
A: There is currently no cure that fixes the underlying genetic mutation permanently. However, the condition is treatable with daily medication that replaces the missing substance the body needs, which allows patients to survive and improves their quality of life.
Q: Can this condition be detected before birth?
A: Yes, if the specific genetic mutation is known in the family, prenatal genetic testing (amniocentesis or chorionic villus sampling) can confirm if the fetus is affected. This allows for treatment to begin immediately at birth.
Q: Does the treatment reverse brain damage?
A: No, the treatment stops the accumulation of toxins and prevents further damage, but it cannot repair brain tissue that has already been destroyed. This is why early diagnosis is critical.
Q: Is it the same as a dietary molybdenum deficiency?
A: No. Dietary molybdenum deficiency is extremely rare and caused by a lack of the mineral in the diet. Molybdenum cofactor deficiency is a genetic inability to use the molybdenum that is already present in the body.
Q: What are the side effects of the treatment?
A: Common side effects of the medication fosdenopterin can include complications related to the IV line (like infection or blockage), fever, and respiratory infections. Doctors monitor these closely.